密度汎関数理論
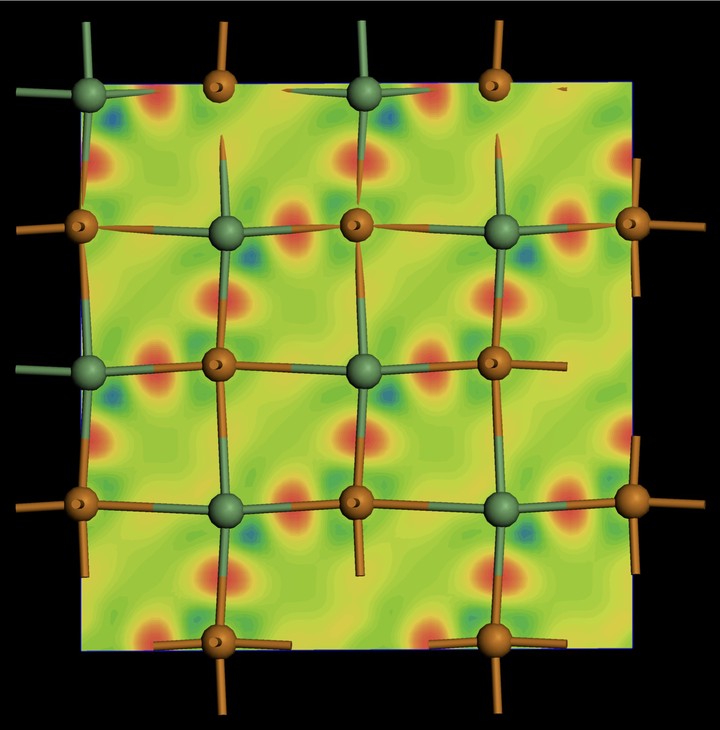 Charge density difference plot for GeTe showing covalent bonds.
Charge density difference plot for GeTe showing covalent bonds.密度汎関数理論(Density-Functional Theory or DFT)は、さまざまな材料特性を予測するために使用されます。 ab-initio分子動力学法を使用して、メルトクエンチシムレーションによってアモルファス構造を生成できます。 平面波DFTソフトであるVASP(Vienna Ab initio Simulation Package)とCASTEP(CAmbridge Serial Total Energy Package)、 そしてFP-LPAW+loであるWien2Kは、構造と電子構造の両方を調査するために使用されます。 X線吸収分光法のデータの解析ために、実空間のグリーン関数ソフトFeff9と FMDNESシミュレーションソフトも利用します。
上の図は、電荷密度差(charge density difference: CDD)を示しています。 短い結合と長い結合の両方を持つGeTe格子の断面を示します。 固体原子と疑似原子の電荷密度の差をとることによって CDDプロットが作成されます。 原子の間に電荷が集まっている場合、Paulingが説明しているように、共有結合を示唆します。