Density-Functional Theory
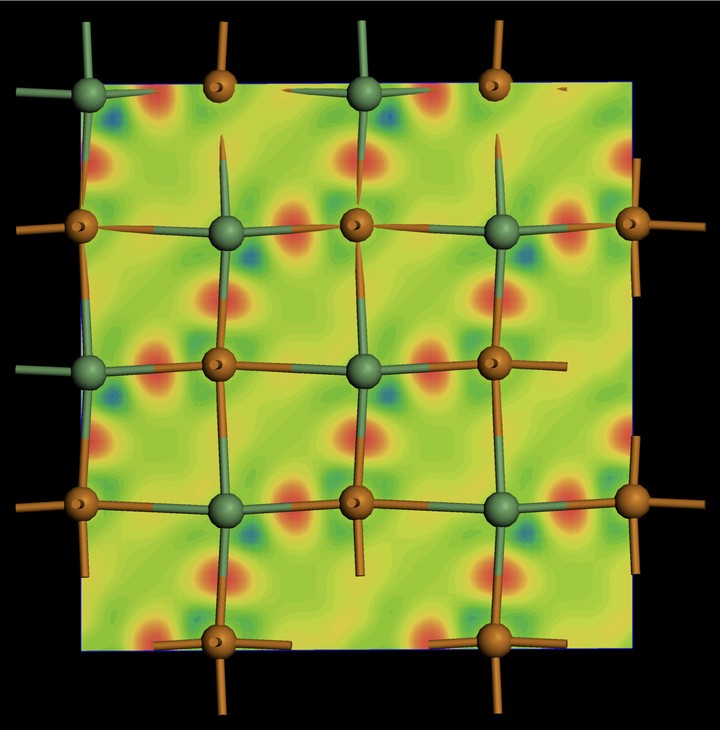 Charge density difference plot for a GeTe. Charge pileup indicates a covalent bond.
Charge density difference plot for a GeTe. Charge pileup indicates a covalent bond.Density-functional theory (DFT) is used to predict a variety of material properties. In addition, ab-initio molecular dynamics based upon DFT is used to examine structural dynamics and to generate metastable (amorphous phases) by means of a melt-quench process. The plane-wave codes VASP (Vienna Ab initio Simulation Package), CASTEP (CAmbridge Serial Total Energy Package), as well as the FP-(L)APW+lo code Wien2K are used to investigate both structural and electronic properties of samples under study. In addition the real-space Greens function code Feff9 and FMDNES codes are used for the simulation of x-ray absorption data. The figure above shows the charge-density difference (CDD) for a cross-section of the GeTe lattice which has both short and long bonds. The CDD plot is constructed by taking the difference between the charge density of the solid and pseudo-atoms. As such the pileup of charge between two atoms is suggestive of a covalent bond as describe by Pauling.
Paul Fons
Professor of Electronics and Electrical Engineering, School of Integrated Design and Engineering
My research interests include the use of computational material science and synchrotron radiation techniques to design and develop new materials.