Machine Learning-based Analysis of ab-initio MD
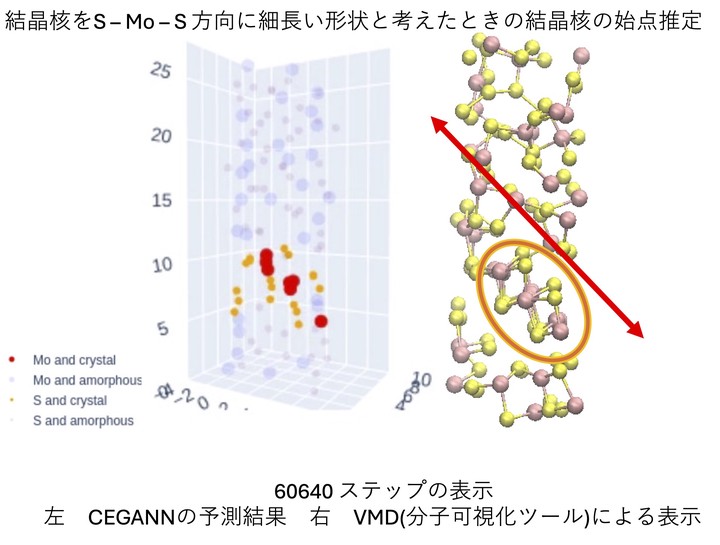 A CEGANN machine learning model identifying sites in a 3D network as being crystalline or amorphous.
A CEGANN machine learning model identifying sites in a 3D network as being crystalline or amorphous.One of the caveats of density-functional theory is that calculations can produce copious amounts of output. Due to the complexity and volume of the output, this can lead to challenges in interpreting the generated output. Here well use CEGANN: Crystal Edge Graph Attention Neural Network for multiscale classification of materials environment[1] In this example, we represent bonds between atoms as well as bond angle information in a mathematical graph in an attention based architecture. These graphs are then introduced as an input layer to a convolutional neural network to “learn” the local environment about atoms in the three-dimensional structure. By training on amorphous and crystalline structures generated using ab-initio* molecular dynamics, the nature of a site in a three-dimensional network can be distinguished as being crystalline or amorphous in nature.
In a thesis, a student used a CEGANN neural network to identify the onset of nucleation in a large scale molecular dynamics run.
[1] S. Banik, D. Dhabal, H. Chan, S. Manna, M. Cherukara, V. Molinero, and S. K. R. S. Sankaranarayanan. Cegann: Crystal edge graph attention neural network for multiscale classification of materials environment. NPJ Computational Materials, 9(1):23, 2023.
Paul Fons
Professor of Electronics and Electrical Engineering, School of Integrated Design and Engineering
My research interests include the use of computational material science and synchrotron radiation techniques to design and develop new materials.